|
顯微鏡為實驗室極重要之工具,尤其是形態學檢查不可或缺的儀器,依其功能及特性可概分為六種,包括 (一) 光學顯微鏡 (二) 暗夜視野顯微鏡 (三) 位相差顯微鏡 (四) 偏光顯微鏡 (五) 螢光顯微鏡 (六) 電子顯微鏡,詳述如下:
|
| 13.1. |
光學顯微鏡 |
| |
光學顯微鏡是組織病理實驗室中極為重要儀器之一,因係精密儀器故操作時需細心,不用時需蓋好以避免灰塵污染,若無特製封套,一般的塑膠袋亦可取用。顯微鏡室必需時時除濕保持乾燥, 以免鏡頭發霉。 |
| 13.1. |
1.顯微鏡基本結構: ( 見圖一 ) |
| |
| (1) |
骨架 ( 支持系統 ):
| 1. |
基座底盤 (Base):支持顯微鏡各部結構。 |
| 2. |
載物臺 (Stage):放置玻片用、附玻片夾可固定玻片及任意調整方向。載物臺中央有圓孔,下方連接集光器,集光器可聚集光線,由此圓孔通過。 |
| 3. |
鏡柱 (Pillar):支持鏡臂及載物臺。 |
| 4. |
鏡臂 (Arm):與鏡柱相聯而呈稍彎之握手處,支持鏡筒及旋轉盤。 |
| 5. |
鏡筒 (Body tube):內含透鏡系統,若鬆動其底部之固定螺絲,可使鏡筒在平面上作360℃ 的旋轉。 |
| 6. |
旋轉盤 (Revolving nosepiece):接於鏡臂末端之下方,可作 360℃ 的旋轉。 |
|
| (2) |
透鏡系統:
| 1. |
接目鏡 (Ocular;Eyepiece):
裝於鏡筒之上端,通常為 10X、15X |
| 2. |
接物鏡 (Objective):
裝於旋轉盤上,通常有 5 個,分別為 4X、10X、20X、40X 及 100X。 |
| 3. |
校光系統:
聚光器 (Condenser):
位於載物台下,由多數透鏡所組成,用於集中由反光鏡反射來的平行光線,使其照射於標本玻片上。
照明器 (Illuminator)
位於集光器下方,由一個 6V,5W 的燈泡以及一個反光鏡組成。
調節輪 (Adjustment)
分粗細調節輪兩種於鏡柱上,旋轉時可使載物台上下移動,具調節焦距的功能。 |
|
|
| 13.1. |
2. 使用顯微鏡應注意事項: |
| |
| 1. |
盡量不要任意搬動顯微鏡,若需移動時必須一手緊握鏡臂,一手托住鏡座,忌單手提,以免因傾斜而造成目鏡與反光鏡滑落破損。放置時亦需小心輕放勿使受強力震動。 |
| 2. |
顯微鏡應放置於使用者之左前方,鏡體應垂直以免傾斜時玻片墜落破損,或觀察潮灦標本玻片時,水流入鏡體內。 |
| 3. |
顯微鏡應經常保持清潔勿使受潮或沾染灰塵,平時應常用絨布清潔鏡頭以外之其他部分。 |
| 4. |
反光鏡、接物鏡、接目鏡等,以拭鏡紙輕輕以圓形方向擦拭鏡面,切勿使用濾紙或其他紙張擦拭鏡頭。 |
| 5. |
配製清潔溶液,定期以拭鏡紙沾該溶液清潔鏡體、鏡片上之污漬及油鏡,切忌用酒精。( 清潔鏡片之清潔液配置法:100% 酒精加乙醚以 1:3 之比例混合 )。 |
| 6. |
使用顯微鏡時,必須坐正而座位高度適宜,觀察時兩眼同時張開,而以任一眼睛觀看,可免眼睛疲勞。 |
| 7. |
用畢顯微鏡後,將載物台移至最低處,並將低倍物鏡旋至對準中央圓孔處。 |
| 8. |
定期請該顯微鏡廠商之專業工程師,給與定期校正及維修,遇有故障切忌自行拆修儀器。 |
|
| |
|
| 13.2. |
暗視野顯微鏡 |
| |
此類顯微鏡最主要使用於較一般細菌小之螺旋體菌及未染色活微生物之觀察。其普通光學顯微鏡上加裝特殊暗視野裝置。聚光器下裝一圓盤而隔絕所有直接之光線,其使光線由聚光器中央向周圍繞射。由於物鏡位於光線之黑暗部份而隔絕所有直接照射之光線,使觀察者只能見到散射之光線。使被檢視之微小目標則於黑暗背景下呈現明亮。 |
| |
|
| 13.3. |
偏光顯微鏡 |
| |
於檢體與光源中間裝置偏極光設備,使偏極光通入檢體而進入物鏡,檢體與眼睛間裝上第二偏光濾鏡,由手旋轉之,以消除大部分之光線並使檢體中產生雙折射之成份能被明見。使用於鑑定結晶物之觀察。 |
| |
|
| 13.4. |
螢光顯微鏡 |
| |
螢光顯微鏡基本上仍為光學顯微鏡,裝上特殊之濾光鏡以使染上螢光染料之物體能經由紫外線照射而呈現。光能為具高能量之紫外線,通常使用高壓之汞蒸氣燈,顯微鏡必需裝置激發濾光鏡,使通過聚光器之光波能選擇最具激發之波長以激發檢體之螢光色素,此外,顯微鏡必須於目鏡與觀察者間裝置隔離濾光鏡,使不需要之光波被吸收而只讓被檢體激發之螢光波長通過,其亦可保護觀察者之眼睛免於受紫外線之照射而損壞。
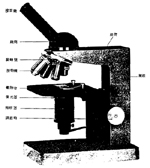
圖 1 光學顯微鏡之基本結構 |
|
|
電子顯微鏡檢查法
|
| 黎南榮 |
| 1. |
概論 |
| |
自古以來,人類對於微小的世界,就一直存有相當的崇敬與好奇心。最初的顯微鏡為放大鏡,也就是單一的凸透鏡。一般考古學家咸信遠在紀元前三千年的兩河流域已有玻璃透鏡的製造。複式顯微鏡以兩個以上的透鏡組合而成。可能是由荷蘭的 Hans, Zacharias Jansen 父子於 1590 年代所發明。而第一架實用性的光學顯微鏡則由荷蘭的 Antonie Van Leeumenhoek 於 1673 年發明,自此光學顯微鏡已帶領人類遊歷了肉眼無法見到的世界,也是使醫學或為科學與藝術的最重要一種工具。顯微鏡其解析力之公式 d = 0.61π/ NA,d 為解析力,π 為波長,NA 為光口角,NA 以油鏡最佳值約 1.4,故光學顯微鏡解像力之極限以自然光時為 0.25μ (250nm) 以單色 400nm 之光源時為 0.17μ。以光學顯微鏡無法區分小於 0.25μ 大小之結構。為了提高解析力以縮短光源之波長為最有效之方法,因而發明了紫外線顯微鏡及x光顯微鏡。為了發明此兩種顯微鏡投資了極大的財力與技術,但所提昇之效果極為有限,兩者的解析力之極限均為 0.1μ。 |
| |
一般光學顯微鏡受限於光波之波長其解析力之極限約為 0.2μ,無法滿足人類永無止鏡的探索欲望,因而才有電子顯微鏡的產生。電子顯微鏡的理論起源於 1920 年代 de Broglie 氏首先發表之電子波動說,電磁波之特性與光波極為類似,但波長很短,電磁波波長之公式為  ,λ 為波長,V 為加速電壓,當加速電壓為 50,000V 時波長約為 0.05nm,但因電磁鏡及 NA 之影響,其解析力僅可達 0.25nm。第一架實用電子顯微鏡則由德國 Ruska 於 1933 年發明,而第一架商用電顯由德國的 Siemens 公司於 1939 年生產。電顯的應用,使科學研究的領域提昇至超顯微的世界,可說是 20 世紀生物界、醫學界及科學界最重要的一種發明。也使 Ruska 先生於 1986 年得到諾具爾物理獎。電子顯微鏡的成像原理與光學顯微鏡一致。主要差別是以電磁波取代光波,電顯內部須維持高度真空狀態,附近最好沒有干擾性的磁場。過去幾十年電子顯微鏡已成為病毒學、生物學、超微結構研究的一種必要工具。 ,λ 為波長,V 為加速電壓,當加速電壓為 50,000V 時波長約為 0.05nm,但因電磁鏡及 NA 之影響,其解析力僅可達 0.25nm。第一架實用電子顯微鏡則由德國 Ruska 於 1933 年發明,而第一架商用電顯由德國的 Siemens 公司於 1939 年生產。電顯的應用,使科學研究的領域提昇至超顯微的世界,可說是 20 世紀生物界、醫學界及科學界最重要的一種發明。也使 Ruska 先生於 1986 年得到諾具爾物理獎。電子顯微鏡的成像原理與光學顯微鏡一致。主要差別是以電磁波取代光波,電顯內部須維持高度真空狀態,附近最好沒有干擾性的磁場。過去幾十年電子顯微鏡已成為病毒學、生物學、超微結構研究的一種必要工具。 |
| |
|
| 2. |
穿透式電子顯微鏡之操作與校正 |
| |
電子顯微鏡由於構造複雜,操作上亦較一般光學顯微鏡困難,因此除了熟練的操作技巧外,亦須具備精確的校正能力,不論是光學顯微鏡的玻璃透鏡或是電子顯微鏡電磁透鏡,由於製造上不可能達到完美,因此會有像差的產生,其校正方法可用較小的孔徑遮去邊緣的電子而改善,但是會使解像力變差。由於磁場的不對稱使電子進入透鏡時在不同軸面之偏轉程度不同而使影像變形,此時可利用像散校正器加強磁場較弱的軸面使之對稱。一個電子顯微鏡的操作員必須知道如何校正像差方可得到較高之解像力,現將基本操作與校正步驟分述如下: |
| |
|
| 2.1. |
開機 |
| |
首先必須打開電源開關及冷卻水系統,然後啟動真空系統,先以迴轉式唧筒抽至 10-2 torr,再以擴散式唧筒抽至 10-4 ~ 10-6 torr 方可使用。現代的電子顯微鏡均設計成全自動真空系統,無需人為操作,可避免人為疏忽而造成油氣倒灌,通常約 20 分鏡可完成。 |
| |
|
| 2.2. |
電子槍的操作與校正 |
| |
當真空度到達後,便可打開電路系統。首先將加速電壓加至 60 到 100 KV,待電壓穩定後再將燈絲加熱至飽和點。燈絲之壽命與真空度及加熱時之溫度有關,最理想之狀況可維持 40 至 80 小時,所謂燈絲飽和點即當燈絲逐漸加熱時在螢光板上可見燈絲之影像,隨著溫度逐漸加高影像由中空環形狀逐漸成一實心光點,此即為燈絲之飽和點,燈絲位置之正確與否可由未達飽和點之光環形狀及在螢光板上可見燈絲之影像,隨著溫度逐漸加高影像由中空環形狀逐漸成一實心光點,此即為燈絲之飽和點,燈絲位置之正確與否可由未達飽和點之光環形狀及在螢光板上之位置來判定,並加以校正,使其在螢光板中央形成一對稱的光環再加熱至飽和點即可。 |
| |
|
| 2.3. |
聚光鏡與可動孔徑之校正 |
| |
聚光鏡可將電子束聚集於標本上,並可調整照明範圍之大小與亮度,通常有一像散校正器可校正磁場之對稱性使電子束成一對稱光點,可變孔徑亦須校正,在選定適當大小之孔徑後,調整孔徑位置使其在焦前 (under focus) 或焦後 (over focus) 時的照明光點均在同一位置形成同心圓。 |
| |
|
| 2.4. |
標本置換與物鏡之校正 |
| |
標本之置換因廠牌與機型而有很大之差異,只須依使用說明書操作即可。物鏡之校正可分為三部份: |
| 2.4. |
1.電流中心與電壓中心 |
| |
電流中心與電壓中心校正乃指一定頻率來回改變物鏡之電流強度或電子槍加速電壓之大小,從影像晃動的方式來判定及校正電子束與鏡柱主軸之對稱性,當電子束與鏡柱主軸一致時,電壓中心可觀察到物體影像以螢光板中央為中心,隨電壓改變的頻率對稱的放大與縮小。而由電流中心則可觀察到影像對稱於中心點沿逆時針與順時針方向來回晃動。若影像晃動不動稱時,可調整電子束傾斜 (beam tilt) 控制鈕之對稱。 |
| 2.4. |
2.物鏡可變孔徑之選擇與調壓 |
| |
物鏡可變孔徑之大小與影像對比度與解像力有關;孔徑越小時對比度越高,解像力越差,因此選擇適當大小的物鏡孔徑可提昇影像之清晰度。孔徑必須調整至主軸中央才不致影響影像品質。 |
| 2.4. |
3.物鏡像散之校正 |
| |
物鏡是檢體呈像最直接的透鏡,也是影響最大的透鏡,而在各種像差影響中,又以像散最為重要,尤其在高倍率的時候,更為重要。物鏡像散之校正以多孔膜 (holly film) 作為校正之樣品,先在高倍下找到一小圓形小洞,調至焦後 (over focus) 使小洞邊緣出現一黑圈,調整像散校正器使黑圈對稱,然後逐漸改變焦距,使小洞邊緣之黑圈消失,若黑圈不同時消失,調整像散校正器使之對稱,如此來回調整直到正焦 (just focus) 時,黑圈均勻消失為止。 |
| |
|
| 2.5. |
對焦與照相 |
| |
經過上述的校正後,只要對焦正確就可得到清楚的影像。通常在正焦時影像之解像力最高,但對比度較差;錯焦 (defocus) 時則會損失解像力,但可提高對比度。通常我們以此微的焦前作為最後對焦狀況,因為此時物體所呈現之影像有較高的對比度,又不會造成影像誤判,尤其是在生物標本的觀察上,對比度的因素遠比解像力重要,故輕微焦前為最理想之結果。
由於電子顯微鏡有相當長的焦深 (depth of focus),螢光板與底片雖不在同一平面上,但對影像之清晰度完全無影響,也就是說只要在螢光板上對焦正確,底片上可得相同清晰之影像,故對焦完成後,調整適當亮度與曝光時間即可將螢光板移開,讓影像直接顯現於底片上。曝光完成後經過負片顯像及相片沖印即可得到永久的影像。 |
| |
|
| 3. |
負染色 |
| |
穿透式電子顯微鏡在生物標本製作技術上,除了可以超薄切片來觀察物體內部的微細結構外,對於一些體積甚小的細菌,濾過性病毒及胞器如胞膜、粒線體、核糖體、蛋白質、核酸等均可用負染色 (negative staining) 直接觀察物體的形狀與大小。以超薄切片方式觀察生物樣品,所得到的解像力約只有 20A°,而利用重金屬物質來增強對比度的負染色可使解像力有效的提昇至 5~10A° 以下,而且樣品製作技術較超薄切片簡單。
負染色是讓重金屬沈積於物體外圍,使物體與其背景呈現強型的對比度。由於生物體組成元素多為分子量較小之碳、氫、氧、氮等,因此電子穿透能力較強,而沈積於外圍之重金屬有較大的原子量對於電子之散射能力較大,電子不易透過,故造成黑白對比與一般切片相反之影像,通常以此法觀察之樣品多為微小顆粒狀結構,如細菌、病毒、粒腺體、蛋白質等,負染色樣品處理較薄切片簡單,花費的時間較短,所需樣品體積很小,卻有較高的解像力與對比度,應用非常普遍。 |
| |
|
| 3.1. |
染劑之配製 |
| |
負染色的染劑種類很多,如磷鎢酸 (Phosphotungstic Acid,PTA),醋酸鈾 (Uranyl acetate),草酸鈾 (Uranyl oxalate),硝酸銀 (Siliver nitrate) 等,只要能溶於水,在電子束照射下穩定及可提供相當對比度的重金屬物質均可作為染劑,其中以磷鎢酸與醋酸鈾最常被使用,配方如下:
| (1) |
1~2% 磷鎢酸,以 NaOH 調整 PH 值至 6.5~8 此染劑相當稱定,可長久保存。 |
| (2) |
0.1~1% 醋酸鈾水溶液,PH 值為 4~5.5 此染劑必須在使用前現配,而且只能在黑暗中保存數小時。
醋酸鈾之顆粒較細,可有較高之解像力,但其對比度較差,且容易滲入物體中而成正染色,磷鎢酸顆粒較粗,但解像力仍可達 10A° 以下,而其對比度高,又不易與物體作用,為相當理想之負染色劑。 |
|
| |
|
| 3.2. |
支持膜之製作及強化 |
| |
由於負染色的樣品均為分散的顆粒,必須要有支持膜方可附著。取 300~400 mesh 之銅網以 Formar 或 Colloidin 做一塑膠支持膜。為了使支持膜有較好之穩定性,在高倍率觀察時不致因電子束照射而破裂,通常在支持膜上再蒸鍍一層約 5mm 厚之碳膜來強化結構,茲將支持膜及碳膜之製造法描述如下: |
| 3.2. |
1. 支持膜:塑膠支持膜製作法可分為滴展法 (drop method),與玻片法 (Slide method),滴展法較簡單,但作出來的支持膜較差,玻片法較困難,但可得品質較佳的薄膜,現以玻片法製作弗氏 (Formvar) 支持膜為例說明支持膜製作法:
| (1) |
取一未曾使用過的玻片,以紗布在拭乾淨 ( 勿用任何液體清洗 ),將一端浸入 0.25 % 的弗氏劑溶液中 ( 將 Formvar 溶於 1.2-dichloroethane 中成 0.2~0.5% 的濃度 )。隨即取出並直立於無塵處風乾。 |
| (2) |
風乾後以刀片沿玻片邊緣刮動數次。 |
| (3) |
在一裝滿蒸餾水之玻璃皿中 ( 水面滿過容器 ),以一乾淨玻棒橫過水面,除去水面灰塵。 |
| (4) |
將玻片以 30 度角緩緩壓入水中使塑膠膜漂浮於水上。 |
| (5) |
以鑷子夾取銅網,粗糙面朝下置於支持膜上,並以攝子尖端輕壓銅網使之與膜密和。 |
| (6) |
以一乾淨玻片由上往下將銅網與膠膜壓入水中再取出風乾即成。
製作支持膜時應避免對著玻璃吹氣,否則膜上會出現許多小孔。 |
|
| 3.2. |
2. 碳膜蒸鍍:碳膜之製作需利用真空蒸鍍機 (Vacum evaporator) 在高真空下以強大電流碳棒產生高熱而蒸著於覆有塑膠支持膜之銅網上。其操作方法如下:
| (1) |
打開真空閥,使玻璃鐘罩內恢復大氣壓。 |
| (2) |
在鍍碳的電極上將削好的之碳棒裝上去,並將覆有支持膜之銅網置於碳棒下方約 10~15 公分處。 |
| (3) |
關啟各開關順序啟動真空系使鐘罩內真空達備而 5╳10-5 torr 以上。 |
| (4) |
將遮板置於碳棒與銅網之間,以避免剛開始加熱碳棒時,不穩定的碳顆粒污染支持膜。 |
| (5) |
逐漸加熱碳棒至尖端紅熱,並可見碳棒有蒸散現像為止。 |
| (6) |
移開遮板,蒸鍍約 30 秒至 1 分鐘,即可關閉電壓電流。 |
| (7) |
關閉及打開適當真空閥,取出樣品。 |
| (8) |
使用完閉後應使操作室保持真空,並打開迴轉式與外界開關後方可關機。 |
|
| 3.2. |
3. 樣品之處理與染色 |
| |
將樣品附著於支持膜上並加以染色的方法有噴灑法 (spray method),滴染法 (drop method) 以及漂染法 (float method) 等,其中以滴染法最為方便,也最常為各實驗室使用,現將步驟條例如下:
| (1) |
準備染劑,濾紙,吸管,自夾式鑷子 (Self-Clamping forceps),樣品 (107 /ml 濃度以上 ) 及覆有支持膜並經膜並經過碳膜強化之銅網。 |
| (2) |
以自夾式鑷子夾住銅網邊緣,將銅網之膜面朝上,滴一滴樣品於其上。 |
| (3) |
靜置數秒至數分鐘後,以濾紙自邊緣吸取液體。 |
| (4) |
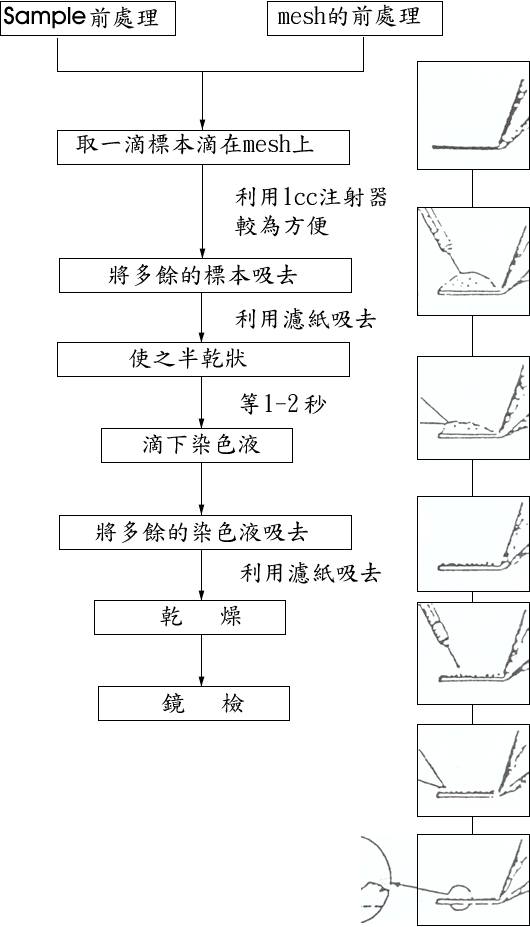
滴上染劑經數秒至半分鐘後以濾紙吸除染劑,風乾後即可觀察。 負染色之程序如附圖
-負染色法的程序
|
|
| |
|
| 4. |
固定與包埋 |
| |
生物樣品通常含有相當多的水分,因此在能夠以顯微鏡觀察微細構造之前,必須經過極複雜的前處理步驟。這些步驟包括固定 (fixation)、脫水 (dehydration)、滲透 (infiltration)、包埋 (embedding)、切片 (sectioning) 與染色 (staining)。 |
| |
|
| 4.1. |
固定的目的與條件 |
| |
樣品固定的最主要目的在於保持其原有的微細構造,避免因接下來的脫水、包埋、切片、染色等樣品處理過程以及於電子顯微鏡中觀察時電子束的照射等行為使樣品發生變形,以致影響影像之判斷。因此,固定時任何足以影響結果的因素都必須加以考慮。通常影響樣品固定品質好壞的原因如下: |
| 4.1. |
1. 固定液的酸鹼度 |
| |
生物細胞均有其特定之 PH 值,當固定液之酸鹼度與細胞相差太大時容易造成細胞內部微細構造的破壞,因此固定液的配製通常以緩衝溶液 (buffer) 來維持一定的 PH 值。動物細胞之 PH 值約為 7.0~7.4,植物細胞為 6.8~7.2,原生動物、無脊椎動物以及柸胎組織為 8.0 左右。 |
| 4.1. |
2. 緩衝溶液的種類及滲透壓 |
| |
對於一般生物組織細胞而言,最常被用來作為配製固定液的緩衝溶液為磷酸鹽衝液 (phosphate buffer) 與二甲胂緩衝液 (cacodylate buffer),有些特殊樣品如組織培養之細胞或分離的白血球、血小板等則視其特性以特殊緩衝溶液為之。除了 PH 值外,滲透壓 (osmolarity) 亦為重要因素,常以蔗糖 (sucrose) 作為維持滲透壓的物質,以避免在固定時造成細胞皺縮或脹大。緩衝溶液的配方如下:
| (1) |
0.2M 磷酸緩衝液 (Phosphate Buffer)
| A 溶液:NaH2PO4.H2O |
27.6 g/L |
| B 溶液:Na2HPO4 ( 不含結晶水 ) |
28.4g/L |
| 23ML 的 A溶液 與 77ML 的 B 溶液混合 (PH=7.3) |
|
| (2) |
0.2M 二甲胂酸緩衝液 (Cacodylate Buffer)
| 二甲胂酸鈉 (sodium cacodylate) ( 不含結晶水 ) |
4.28g |
| 蒸餾水 |
100 mL |
| 以 1N 的 HCl 調整 PH 值至 7.0~7.3 |
|
|
| 4.1. |
3. 固定時間的長短 |
| |
由於多數固定液滲透的速度不快,故需固定一段時間,然而固定時間長極易造成細胞中物質的流失,因此,固定時間應儘可能縮短,通常在數小時中即應完成。當然,固定時間的長短,與樣品之大小、固定液的種類、濃度均有關聯,如果是單細胞生物或組織培養的單層細胞,固定時間只須 15 分鐘即可。 |
| 4.1. |
4. 固定時的溫度 |
| |
習慣上,樣品的固定多在 0°~4℃ 中進行,其目的在減緩細胞中酵素的活性,以避免自溶作用 (autolysis) 的產生。自溶的現像在動物細胞中最為明顯,當細胞死亡時,存在於溶素體 (lysosome) 中的酵素會釋出,將細胞質內的成分或胞器瓦解,造成細胞中出現殘缺的胞器可使細胞的破壞減至最低。不過高溫可使化學反應加速,在高溫下固定液的滲透作用也較快,例如對於細菌的內孢子,在 40℃ 下固定可得到較好的結果。 |
| 4.1. |
5. 樣品體積的大小 |
| |
為了使固定的時間縮短以及將來滲透包埋的方便,樣品的體積不宜過大,通常最適當的大小為 0.5~1mm 左右,如此大小的樣品可在一個小時中完全固定。
一個理想固定的生物樣品在穿透式電子顯微鏡下,細胞內如細胞膜 (plasma membrane)、內質膜 (endoplasmic reticulum)、高爾基體 (Golgi complex)以及核膜 (nulear membrane) 等膜狀構造都應保持連續而完整,高倍率下並可觀察到脂雙層 (lipid bilayer) 結構,粒腺體 (mitochondrion) 與葉綠體 (chloroplast) 也應保持完整而無脹大或皺縮現象,至於細胞質 (cytoplasm) 與核質 (nucleoplasm) 則應呈現緻密而無不規則空泡存生。 |
| |
|
| 4.2. |
固定液 |
| |
在電顯技術上最常作為固定劑來使用的物質為醛類 (aldehyde) 與四氧化鋨 (osmium tetroxide)。醛類包括甲醛 (formaldehyde)、戊二醛 (glutaraldehyde) 以及丙烯醛 (acrolein) 等,主要在固定細胞中的蛋白質,其滲透能力比四氧化鋨快速;四氧化鋨的作用在於與不飽和脂肪酸結合,適合胞膜的固定,且因其具有原子量甚大的鋨,故同時兼有染色的功能。
在三種醛類中,以戊二醛 (glutaraldehyde) 最常被使用,固定效果也最佳,分子結構為
其固定的方式為與蛋白質分子形成交錯結合,由於戊二醛具有兩個醛基 (aldehyde group),可經由醛醇縮合作用 (aldol condensation) 將兩個或三個戊二醛結合成一較大的分子,然後再與蛋白質形成鍵結。四氧化鋨 (OsO4) 的分子量為 254.2,熔點 41°C,在室溫下為一略帶黃色的固體,由於其兼具固定與染色的雙重作用,在生物樣品的處理上極為重要。OsO4 的固定作用在於能與超有雙鍵的不飽和脂肪酸形成雙脂鍵結 (diester crosslinkage),對於細胞中膜狀構造的穩定最有效用。
配製戊二醛 (glutaraldehyde) 與 OsO4 可直接購買 5ml 或 10ml 裝的 25% 戊二醛與 4% 的 OsO4,再以適當的緩衝溶液分吸配製成 2.5% 與 1% 即可。由於此等固定液對人體有害,尤其是 OsO4 之蒸氣,可破壞眼、鼻及口腔內的皮膜細胞,最好在抽氧罩中操作,並避免直接接觸皮膚上。另外在戊二醛中加入 1%~4% 的丹寧酸 (tannic acid),可使胞膜及微小管的構造更為明顯。
化學固定可利用浸泡 (immersion) 或灌流 (perfusion) 的方式進行。浸泡方式較簡便,只須將欲固定之組織或器官取出,切成適當大小後浸於固定液中即成,但固定效果較不如灌流方式完全。固定前應前準備蠟板、竹籤、小玻璃瓶 (vial)、固定液、雙面刮鬍刀片以及冰塊等,首先將預冷的固定液滴於蠟上,在殺死動物後迅速取出所要之組織,先以刀口相對的方式將組織在固定液中切成 1mm3 大小,再以竹籤挑選適當大小之組織置於小玻璃瓶內,並在冰浴中進行固定。灌流固定則必須準備大量固定液,麻醉動物後由血管中灌注,此法常用於不易在短時間內取出之組織器官上,如腦、脊髓等。化學固定常以雙重固定為之,初固定 (pre-fixation) 以戊二醛而後固定 (post-fixation) 則以 OsO4 進行。在兩次固定之間以及固定液,然後再作脫水步驟。習慣上初固定常在 0~4℃ 的冰浴中進行;以避免因自溶作用而使細胞中之微細構造破壞,後固定則可在室溫下進行。 |
| |
|
| 4.3. |
脫水 |
| |
由於多數包埋劑均無法與水互溶,因此在包埋之前必須先經過脫水過程,使組織中的水分由其他可與包埋劑互溶的有機溶劑取代。最常用來作為脫水劑的物質為酒精 (ethanol) 與丙酮 (acetone),其步驟為將固定完成之組織浸泡於一系列濃度 (50%、70%、80%、90%、95%、100%) 的酒精或丙酮中各 10 至 15 分鐘,即完成脫水過程。為了達到確實脫水的目的,在 100% 的酒精或丙酮中可事先加入無水硫酸銅粉末 (CuSO4 ),以確保脫水劑的純度。
脫水劑的濃度差距不宜過大,時間不宜過長,否則極易造成細胞皺縮變形或細胞內物質流失,更換不同濃度的脫水劑時,應避免讓組織暴露在空氣中而造成乾燥。 |
| |
|
| 4.4. |
包埋劑 |
| |
電子的穿透能力很差,樣品必須較 0.1μ 為薄方可讓電子穿透,一般光學顯微鏡技術上使用蠟作為包埋劑,其硬度不夠,無法切出 1μ以下厚度的切片,不適合作為電顯技術使用。要成為理想的包埋劑必須具備以下幾個條件:(1) 黏稠度低,滲透容易;(2) 聚合均勻,不致造成樣品傷害;(3) 硬度適當,易切薄片;(4) 在電子束的照射下穩定,不變性。最早被用於電子顯微鏡技術上之包埋劑為甲基丙烯酸 (methacrylate),但因其聚合時易造成皺縮及樣品傷害,在電子束照射下又易揮發,不甚理想。至 1960 年代已逐漸被環氧樹脂 (epoxy resins) 所取代。環氣樹脂為一些黏稠度相當大的樹脂 (resin),在聚合時常有加速劑 (accelerator) 來促成聚合的產生,有硬化劑 (hardener) 來強化所形成的交錯結合,以及可塑劑 (plasticizer) 來調整其硬度,較常被使用之樹脂種類及配方如下:
| (1) |
Epon
| |
A |
B |
| Epon 812 |
62 ml |
100 ml |
| DDSA |
100 ml |
---- |
| NMA |
---- |
89 ml |
| DMP-30 |
|
|
配置 Epon 時以 A 溶液 7mL、B 溶液 3 mL、DMP-30 0.15mL 的比例混合即成,其硬度可由 A、B 之比例調整,B 溶液越多則越硬。常以氧化丙烯 (propylene oxide) 作為過渡溶液,聚合時應在 60℃ 烘箱中烘烤 48 至 72 小時。 |
| (2) |
Surr's resin
| ERL-4206 |
10.0 g |
| NSA |
26.0 g |
| DER-736 |
6.0 g |
| DMAE |
0.4 g |
Spurr's resin 之黏稠度較低,容易滲透,其硬度由 DER-736 控制,量多時較軟,量少時變硬,可利用酒精或丙酮作為過渡溶液,通常在 70℃ 烘箱中置放 8 至 48 小時即可完全聚合。
多數包埋劑均為有害人體物質,使用時應格外小心,廢棄或殘餘之包埋劑應置烘箱中聚合硬化,以免造成污染。 |
|
| |
|
| 4.5. |
滲透與包埋 |
| |
電子顯微鏡技術上所使用之包埋劑黏稠度都很大,因而滲透效果的好壞也成為樣品處理的重要因素之一。為使包埋劑能順利透入組織細胞中,通常以一定比例之過渡溶劑 (transitional solvent) 與之混合,將完成脫水步驟的組織塊浸泡其中,並逐漸增加包埋劑之比例至 100% 為止。過渡溶劑為能與包埋劑完全互溶之有機溶液,如酒精、丙酮或環氧丙烷 (propylene oxide) 等,滲透時先將過渡溶劑與包埋劑以1:1的比例混合均勻,把組織塊浸泡其中,再以 1:3 乃至純的包埋劑分別進行滲透,時間長短視所選用之包埋劑種類及組織塊緻密度而定,通常 1:1 及 1:3 各浸泡 30 分鐘,純的包埋劑則滲透兩次,每次各一小時。滲透時,應經常搖盪組織,可有較好的滲透效果。滲透不完全時常會造成組織細胞中出現不應有之空隙,影響切片與觀察。使用 Epon 作為包埋劑時常以環氧丙烷當過渡溶劑,Spurr's resin 則可與酒精或丙酮互溶。滲透完全後即可進行包埋,步驟如下:
| (1) |
將膠囊 (beem capsules) 烘乾立於包埋架中,以滴管沿膠囊內壁滴一小滴包埋劑,使其慢慢流至底部。 |
| (2) |
以竹籤挑取一適當大小久組織塊輕置膠囊中。 |
| (3) |
沿膠囊內壁加入包埋劑直至八分滿為止。 |
| (4) |
包埋完成置於預先設定溫度之烘箱中進行聚合。
若要觀察特定方向之組織,可用平板包埋 (flat embedding) 方式包埋,至於細小之樣品如孢子、細菌、花粉等,可將之離心後先包埋於洋菜膠 (agar) 中,再進行脫水、滲透與包埋。 |
|
| |
|
| 4.6. |
穿透式電子顯微鏡樣品前處理程序 |
| |
| (1) |
2.5% 戊二醛 (glutaraldehyde)/0.1M
緩衝溶液 (buffer)(0~4℃) |
1~2hr
|
| (2) |
0.1M 緩衝溶液 /5% 蔗糖 (0~4℃) |
15min
|
| (3) |
0.1M 緩衝溶液 /5% 蔗糖 (0~4℃) |
15min
|
| (4) |
1% 鋨 (osmium) /0.1M 緩衝溶液 ( 此步驟以後可在室溫下進行 ) |
1hr
|
| (5) |
0.1M 緩衝溶液 /5% 蔗糖 (0~4℃) |
15min
|
| (6) |
0.1M 緩衝溶液 /5% 蔗糖 (0~4℃) |
15min
|
| (7) |
50% 酒精 |
10min
|
| (8) |
70% 酒精 |
10min
|
| (9) |
80% 酒精 |
10min
|
| (10) |
90% 酒精 |
10min
|
| (11) |
95% 酒精 |
10min
|
| (12) |
100% 酒精 |
15min
|
| (13) |
100% 酒精 |
15min
|
| (14) |
100% 丙酮 |
15min
|
| (15) |
100% 丙酮 |
15min
|
| (16) |
100% 丙酮 /Spurr's resin 1:1 |
30-60min
|
| (17) |
100% 丙酮 /Spurr's resin 1:3 |
30-60min
|
| (18) |
Spurr's resin 1:3 |
1-3 hr
|
| (19) |
Spurr's resin 1:3 |
1-3 hr
|
| (20) |
包埋 |
1-3 hr
|
| (21) |
置於 70℃ 烘箱中 8 至 24 小時 |
|
|
| |
|
| 5. |
超薄切片 |
| |
由於電子的穿透力極低,樣品厚度必須薄至 50 至 100nm,與光學顯微鏡切片的 3-7μ 的厚度相比約只有 1/50 故稱之為超薄切片。理想的超薄切片應當平整而無破洞,刮痕或皺褶;而且每一切片可連接在一起成為一條平直的長帶。超薄切片的製作除了要熟悉並正確的使用超薄切片機外,樣品前處理的好壞,樣品塊修整,玻璃刀及支持膜的製作技術均可影響切片的品質,現分別詳述如下: |
| |
|
| 5.1. |
樣品塊的修整 |
| |
包埋完成後的樣品在切片前必須經過適當的修整,方能切出理想的薄片。習慣上我們將樣品修整成梯形,大小約 0.2~0.5mm 之間,高度不宜超過 0.2mm 否則在切片時易造成震動或斷裂。修整的過程通常在解剖顯徵鏡下進行,首先以單面刀片在樣品塊上作平整的削切,直至露出組織為止,然後再從垂直與水平方向分別切出梯形的兩底及腰。切口應平整而無缺刻,否則切出來的切片便無法連接成帶狀,造成撈片困難。 |
| |
|
| 5.2. |
銅網與支持膜 |
| |
如同光學顯微鏡的標本製作時需要有玻片來承載切片一樣,電子顯微鏡的標本薄片也必須有物體支持方可置於顯微鏡中觀察,由於電子無法穿過玻璃,因此具有許多網孔的銅網便取代了玻片的地位。銅片的網格大小以 "mesh" 來區分,如標示為 200mesh 表示每英吋有 200 個分格,換句話說,mesh 愈大表示網孔徑愈小。銅網有正反面之分,在燈光下呈現較淡顏色且不反光者為粗糙面亦即是正面,是製作支持膜或放置切片的一面;而顏色略深會反光的面是背面。銅網片一般以銅為材料,也可視需要以鎳、銀、黃金或白金取代。習慣上銅網上不重覆使用,因為使用過的銅網片清理起來非常費事,未使用過銅網片只須以酒精或丙酮浸泡數分鐘再經過自然風乾後即可使用。
支持膜為一屬極薄的塑膠膜或炭膜,覆蓋於整個銅網片表面,其作用在使薄切片能平整的附著於銅網片上,使用在電子束的照射下,不易破裂或捲縮,尤其是使用 200mesh 以下的銅網片,通常都必須製作支持膜。至於像病毒,蛋白質分子等微細顆粒標本的觀察則必須完全靠支持膜提供的作用,否則根本無法觀察。支持膜之製作方法,如前章所述。 |
| |
|
| 5.3. |
超薄切片機與玻璃刀 |
| |
超薄切片機,雖有不同廠牌與機型,但其基本構造均大致相同,包括一個可前後左右移動及傾斜、旋轉的刀座,一個可固定組織塊並作上下左右運動的標本臂,一組照明及放大觀用的解剖顯微鏡,及一個利用機械或溫度方式控制切片厚度的控制器,機械推進方式乃靠一組精密的螺紋裝置,在標本臂每上下運動一次時向前推進一小段距離 ( 切片厚度 )。溫度推進方式則利用金屬受熱膨脹的原理,穩定地加溫使標本臂向前推進,切片厚度則依加熱的程度調控,而標本臂運動之速度或頻率也會影響切片厚度。由於超薄切片機對震動和溫度非常敏感,故應安置於穩固或防震的桌面上,並有適當的隔間以避免切片時受到干擾。
超薄切片時所使用的刀子有玻璃刀與鑽石刀兩種,玻璃刀的製作容易且較便宜,用過即可丟棄,故常被使用;鑽石刀的壽命較長,可切較堅硬的樣品,但因價錢昂貴,使用後須妥善保養。玻璃刀的鋒利刀口是利用加壓斷裂玻璃而作,可利用製刀鉗或製刀機製作,首先將玻璃片或玻璃條切割成一英吋大小的正方形。再由對角線切割一裂痕,自兩側及底部加壓使玻璃斷裂為二即成。理想的刀口應呈平整均勻,在解剖顯微鏡下無鋸齒狀缺痕,適合切片的刀口佔整個裂面 1/3至 1/2。 為了使切片容易展開及撈取必須在玻璃刀口周圍做一水槽使切出的薄切片能浮於水面成一長帶。水槽的製作常以鍍有金屬的膠帶為材料,將膠帶沿玻璃刀口後方圈出一船形水槽,並將多餘的部分切除,然後將與玻璃刀接觸面以蠟或指甲油封合。也可買到預先作好大小及形狀的塑膠水槽,只須安裝於刀口上再以蠟或指甲油封合即成。 |
| |
|
| 5.4. |
切片 |
| 5.4. |
1. 樣品塊及刀子的架設 |
| |
依包埋方式 ( 膠囊包埋或平板包埋 ),選用適當的標本固定座,將組織塊固定於切片機的標本臂上並將標本臂退回起點,利用銼刀或安全刀片切削成 0.5 ~ 0.6 ╳ 0.5 ~ 0.9mm 的切削面,組織塊的高度不可超過 6 ~ 7mm 以上。 |
| 5.4. |
2. 厚切片 |
| |
利用玻璃刀作一較厚之切片 (0.2~2μm),滴一滴蒸餾水於玻片上,將切片浮於蒸餾水上,並從玻片下加熱則切片延伸而附著於玻片上,用 Toluidine blue 染後利用光學顯微鏡找出需要的組織部分再加以整修包埋塊。 |
| 5.4. |
3. 薄切片 |
| |
將刀子之水槽加滿蒸餾水並調整燈光角度,設定切片速度於 1mm/sec,切片厚度為 60 至 90nm,即可開始切片,切片的實際厚度以從切片在水面上的顏色來判斷,詳如附表,一般以銀白而略帶金黃色之切片厚度為宜,灰色表示太薄,紫色、藍色則表示太厚,理想的切片應成一顏色均勻的長條帶狀。
|
切片顏色與厚度
|
|
顏 色
|
厚度 (nm)
|
|
灰 色
|
20 以下
|
|
灰銀色
|
60~20
|
|
銀 色
|
90~60
|
|
金 色
|
150~90
|
|
紫 色
|
190~150
|
|
藍 色
|
240~190
|
|
| 5.4. |
4. 薄切片的撈取 |
| |
以一支前端粘有羊眉毛或睫毛的竹籤,以划水的方式將長帶狀切片移至水槽較深處,並以眉毛邊緣輕碰帶狀切片切割處使之斷成五至六片的長度,夾取銅網片,支持膜朝上斜浸入水中,以上接觸第一片切片,並緩緩拉出水面,切片便平整地附於銅網片上,再用濾紙吸去多餘的水分後置於襯有濾紙的培養皿數小時至隔夜,待完全風乾後方可進行染色。 |
| |
|
| 5.5. |
切片時的困擾 |
| |
切片時經常會出現一些狀況而造成切片不順利或於觀察時有許多缺點,若能確實瞭解其原因並作適當修正則可減少時間與金錢的浪費,現將常見的困擾及可能的原因列表如下:
|
切片時常見的困擾及可能的原因
|
|
狀況
|
可能原因
|
切片不連續
( 間隔得到切片 ) |
‧樣品塊未完全穩定地固定於標本臂上 |
| ‧刀子在刀座上鬆動 |
| ‧包埋時硬度不夠 |
| 切片被刀口拖離 |
‧水槽中的水位過高 |
| ‧刀子傾斜角度不對 |
| 切片擠壓皺縮 |
‧水槽中的水位過低 |
| ‧樣品塊硬度不夠 |
| 切片顏色不均 |
‧樣品塊的軟硬不均 |
| ‧包埋不良 |
| 切片無法成帶狀 |
‧樣品塊修整時,梯形的上下底不夠平整 |
| ‧刀口不理想 |
| 帶狀切刀片不直 |
‧修整之梯形上下底不平行 |
| 切片上出現刮痕 |
‧刀口上有異物或刀口有缺刻 |
| ‧樣品塊中有堅硬固體沈積物質 |
| 切片上有平行條紋 |
‧切片機不穩固產生細微的振動 |
| ‧切片速度太快 |
|
| |
|
| 5.6. |
染色 |
| |
在生物電子顯微技術上對比度的考慮非常重要,此乃由於生物標本所組成的成分均為原子量甚小的碳、氫、氧、氮等元素,因而對電子散射的能力較弱,影像之對比度較低,因此,如何有效的增加對比度成為一重要的課題,通常影響對比度有下列數種因素:
| (1) |
切片厚度
當電子穿過物體時,物體的密度越大對比度越高,樣品的厚度增加時,電子穿過其中的路徑變長,造成散射的機會增加,因此,切片厚度與其密度的乘積決定了對比度的高低,理論上厚切片應有較高的對比度,但因受電子穿透力及電顯景深很長,造成微細構造影像重疊,故切片厚度有一定限制,只能以其他方法增強其對比度。 |
| (2) |
染色
染色可說是增強影像對比度最有效的方法,正染色是使重金屬染劑直接與物體的構成分子結合造成電子散射的效果以提高對比度。 |
| (3) |
加速電壓的速度
加速電壓的大小可決定電子運動速度的快慢,使用較低加速電壓觀察樣品時因電子運動速度慢較易產生散射現象,可有較高的對比度。 |
| (4) |
物鏡孔徑的大小
物鏡孔徑的大小與影像對比度及解像力均有關聯,孔徑越小時對比度越高,但解像力越差,要提高對比度只須選用較小的物鏡即可,一般生物樣品均採用較小的物鏡孔鏡以提高其對比度。 |
| (5) |
錯焦程度
通常在正焦時影像之解像力最高,但對比度較差,錯焦時可提高對比度,習慣上以些微的焦前做為對焦狀況。 |
| (6) |
底片的顯像與相紙的選擇
經過曝光的底片可由不同的顯影液及提高其對比度,例如以 Kodak D-19 顯影就比 D-76 有較高的反差,在相片沖印時也可選用不同號數的相紙來提高對比度,一般而言號數越大其對比度越強。 |
| |
上述各種方法以染色來提高對比度的效果最佳,影響也最大,染色的作用是讓電子散射能力較強的金屬與樣品中特定的分子結合,使其呈現較好的對比度,最常用的金屬有鈾 (U) 和鉛 (Pb),其他如鋨 (Os)、銀 (Ag)、鐵 (Fe)、金 (Au) 及鎢 (W) 亦可使用。一般的切片染色常以醋酸鈾 (Uranyl acetate) 及檸檬酸鉛 (lead citrate) 作雙重染色,醋酸鈾能與 RNA、膠原纖維、膜狀構造染色、檸檬酸鉛則可使經過四氧化鋨固定後的細胞膜更清楚,二者配方如下: |
|
| 5.6. |
1. 醋酸鈾 {(CH3COO)3 UO2H2O,Uranyl acetate}
(Watson:J.B.C.C. 4,474,1985)
對於 RNA,膠原纖維,膜狀構造等的染色甚佳。
| (1) |
染色液的配製 |
| |
作 50% Ethyl Alcohol 的飽和溶液 ( 約 13%),或水的飽和溶液 ( 約 8%) 溶解後,過濾數次,放置一天後使用。 |
| (2) |
染色時間:
| 1. |
Epoxy 包埋的切片,在室溫下,染約 20 ~ 25 分。 |
| 2. |
Methacrylate 的切片,則染 10 ~ 15 分。 |
|
| (3) |
清洗: |
| |
染色後用 50% Ethyl alcohol,清洗 3~4 次。 |
| (4) |
使用上須注意:
| 1. |
在室溫下,如長久曝露在光線下,易呈黑色混濁,變成不能使用,故最好保存於陰暗處。 |
| 2. |
如放置過久,易起化學變化,而產生沈澱,染色效果降低,所以宜少量配製,保存期限不宜超過三個月。 |
|
|
| 5.6. |
2. 鉛染色 |
| |
Watson 的染色液,易與二氧化碳作用,不穩定,經 Karnousky,Millonig,Reynolds 等改進的鉛染色液,較常被使用。
硝酸鉛 PB(NO3)2 (Reynold J.C.B. 17.208.1963)
| (1) |
染色液的配製:
| A液 |
硝酸鉛 {Pb(NO3)2}------------------------------------------ |
1.33g |
| |
檸檬酸鈉 (Na3C6H5O7 ‧ 2H2O)--------------------------- |
1.76g |
| |
蒸餾水---------------------------------------------------------- |
30ml |
| B液 |
IN NaOH ------------------------------------------------------- |
8ml |
| A 液經強振盪,使之溶解。呈乳白色,經 30 分鐘後,加 B 液 8ml,液體呈無色透明,再添加蒸餾水至 50ml。 |
|
| (2) |
染色時間:
在室溫下,染約 5~10 分。 |
| (3) |
鉛染色的清洗:
染色後用 0.1N 的 NaOH,快速地清洗一次然後以蒸餾水清洗 3~4 次。 |
| (4) |
使用上須注意:
| 1. |
要注意防止碳酸鉛的形成。 |
| 2. |
使用的試藥純度要高。 |
| 3. |
蒸餾水要使用之前,先煮沸一次,將二氧化碳趕出。 |
| 4. |
染色時,在染色用的培養內,放入 NaOH 顆粒,吸收皿內的二氧化碳,以免碳酸鉛的形成。 |
| 5. |
可在室溫下保存,保存的容器宜採密封且不易搖動者。 |
| 6. |
不要作長時間的染色,否則成像的顆粒變粗。 |
茲將常見之病毒與細菌之電顯圖片表列於後供各位參考。 |
|
| |
|
| |
|
| |
重要參考文獻
| 1. |
陳家全,李家維,楊瑞森。生物電子顯微鏡學,國科會精儀中心,科儀叢書;4,1991. |
| 2. |
蘇加祥,電鏡試樣製作,日製產業株式會社,科學儀器海外部。1980. |
| 3. |
Doane,F W,and Anderson N,Electron microscopy in diagnostic virology. Cambridge university press. 1987. |
| 4. |
Glauerf A M. Practical methods in electron microscopy. Elsevier/North-Holland Biomedical press.1977. |
|